1 差示扫描量热法(DSC) 的工作原理
差示扫描量热法(DSC)是19世纪60年代以后研制出的一种热分析方法,它是在程序控制温度下,测量输入到试样和参比物的功率差与温度的关系的一种技术[1,2]。这种测量差值的变化,其精度要高于对热流或者功率的绝对值的测量[3]。其主要特点是使用的温度范围比较宽(-175~725 ℃)、分辨能力高和灵敏度高。由于仪器能定量地测定各种热力学参数(如热焓、熵和比热等)和动力学参数,所以在应用科学和理论研究中获得广泛的应用。根据测量方法的不同,又分为2种类型:功率补偿型DSC和热流型DSC,2者的最大差别在于结构设计原理上的不同。一般实验条件下,选用的是功率补偿型DSC。该仪器有2只相对独立的测量池,其加热炉中分别装有测试样品和参比物,这2个加热炉具有相同的热容及导热参数,并按相同的温度程序扫描。参比物在所选定的扫描温度范围内不具有任何热效应。因此在测试的过程中记录下的热效应就是由样品的变化引起的。当样品发生放热或吸热变化时,系统将自动调整2个加热炉的加热功率,以补偿样品所发生的热量改变,使样品和参比物的温度始终保持相同,这就是功率补偿DSC仪的工作原理。
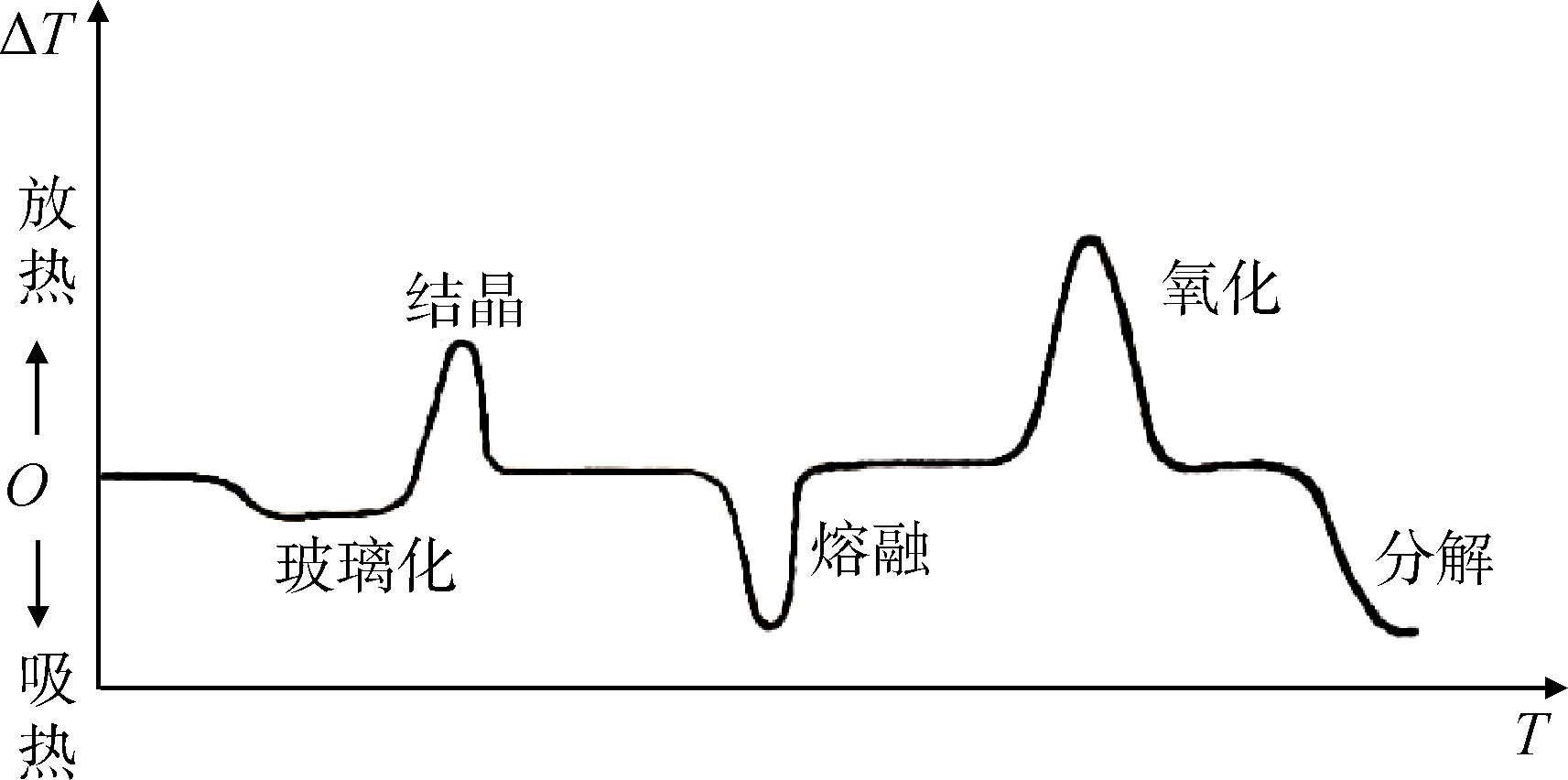
具体实验过程中,以设定的升温速率对试样进行加热,刚开始加热时,试样和参比物以相同温度升温,试样没有热效应,DSC曲线上为平直的基线。当温度上升到试样的玻璃化转变温度时,大分子的链段开始运动。试样的热容发生明显的变化,由于热容增大需要吸收更多的热量,于是DSC曲线上出现一个转折,该转折对应的温度,即玻璃化转变温度(Tg)。若试样是能结晶的并处于过冷的无定形状态,则在玻璃化温度以上的某个温度范围开始结晶,同时放出大量的热量,此时DSC曲线上表现为放热峰;再进一步加热,晶体开始熔融而需要吸收热量,其DSC曲线在放热峰相反的方向出现吸热峰。将熔融峰顶点对应的温度记作熔点(Tm);继续加热试样可能发生其他变化,如氧化、分解(氧化是放热反应,分解是吸热反应,见图1)。因此,根据DSC曲线可以确定高聚物的玻璃化转变和特征温度。
图1 聚合物的DTA和DSC曲线示意图
Fig.1 Diagram of DTA and DSC curves of polymers
2 影响 DSC 实验结果的因素
2.1 升温速率
一般认为DSC 的定量测定主要热力学参数是焓,受升温速率影响很小,但实际测试的结果表明,升温速率太高会引起试样内部温度分布不均匀,炉体和试样也会产生热不平衡状态,所以升温速率的影响很复杂。一般主要影响DSC 的峰值、峰形的大小和窄宽[4]。升温过快,基线漂移明显,峰的分辨率较差,同时峰顶温度会向高温方向偏移;升温速率慢,峰尖锐,因而分辨率也好。升温速率对Tg的测定影响较大,因为玻璃化转变从分子结构来看,是高聚物无定形部分从冻结状态到解冻状态的一种松弛现象[5,6],升温速率太慢,转变不明显,甚至观察不到;升温快,转变明显,但Tg移向高温。升温速率对Tm的影响不大,但有些聚合物在升温过程中会发生重组、晶体完善化,使Tm和结晶度都提高。
2.2 参比物
要得到平稳的基线应尽可能选择与试样的热容、导热系数、粒度等性质比较相近的热惰性物质作为参比物。常用的参比物有氧化铝、煅烧过的MgO和SiO2等。
2.3 气氛和压力
不同气体热导性不同,会影响炉壁和试样之间的热阻,而影响出峰的温度和热焓值。某些样品或其热分解产物可能与周围的气体进行反应,因此应根据需要选择适当的气氛。另一方面,对于释放或吸收气体的反应,出峰的温度和形状还会受到气体压力的影响。气氛可以是静态的,也可以是动态的。就气体的性质而言,可以是惰性的,也可以是参加反应的,视实验要求而定。对于聚合物的玻璃化转变和相转变测定,气氛影响不大,但一般都采用氮气,流量在30 mL/min左右。
2.4 试样用量
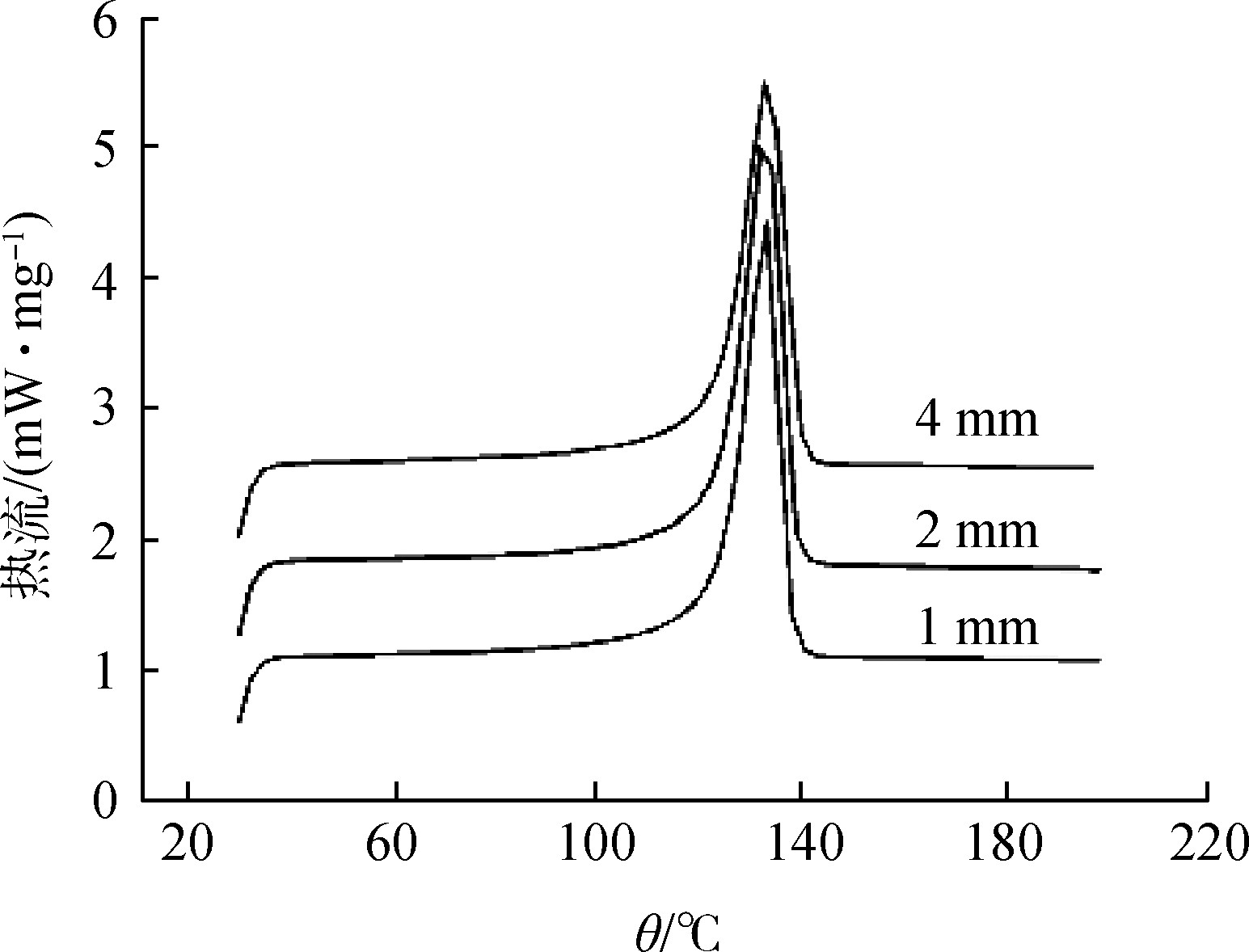
试样量小,峰小而尖锐,峰的分辨率好[7];试样量大,峰大而宽,峰的位置移向高温方向。在仪器灵敏度许可的情况下,试样应尽可能的少,不可过多,以免使其内部传热慢、温度梯度大而使峰形扩大和分辨率下降。有人曾研究了高密度聚乙烯(HDPE)的试样用量对DSC的影响[8],实验结果见图2。
2.5 试样粒度
粉末粒度不同时,由于传热和扩散的影响,会出现试验结果的差别。通常粒度越细,出峰温度降低,峰宽变小。但是其相应的热反应是不变的,只是反应速度有变化。粒度过细时,由于失水很快,也会影响曲线形状。图3是HDPE的试样粒度对DSC的影响[8]。
图2 样品的质量对DSC测试的影响
Fig.2 Effect of sample quality on DSC testing
图3 样品的粒径对DSC测试的影响
Fig.3 Effect of sample size on DSC testing
3 DSC在高分子中的应用
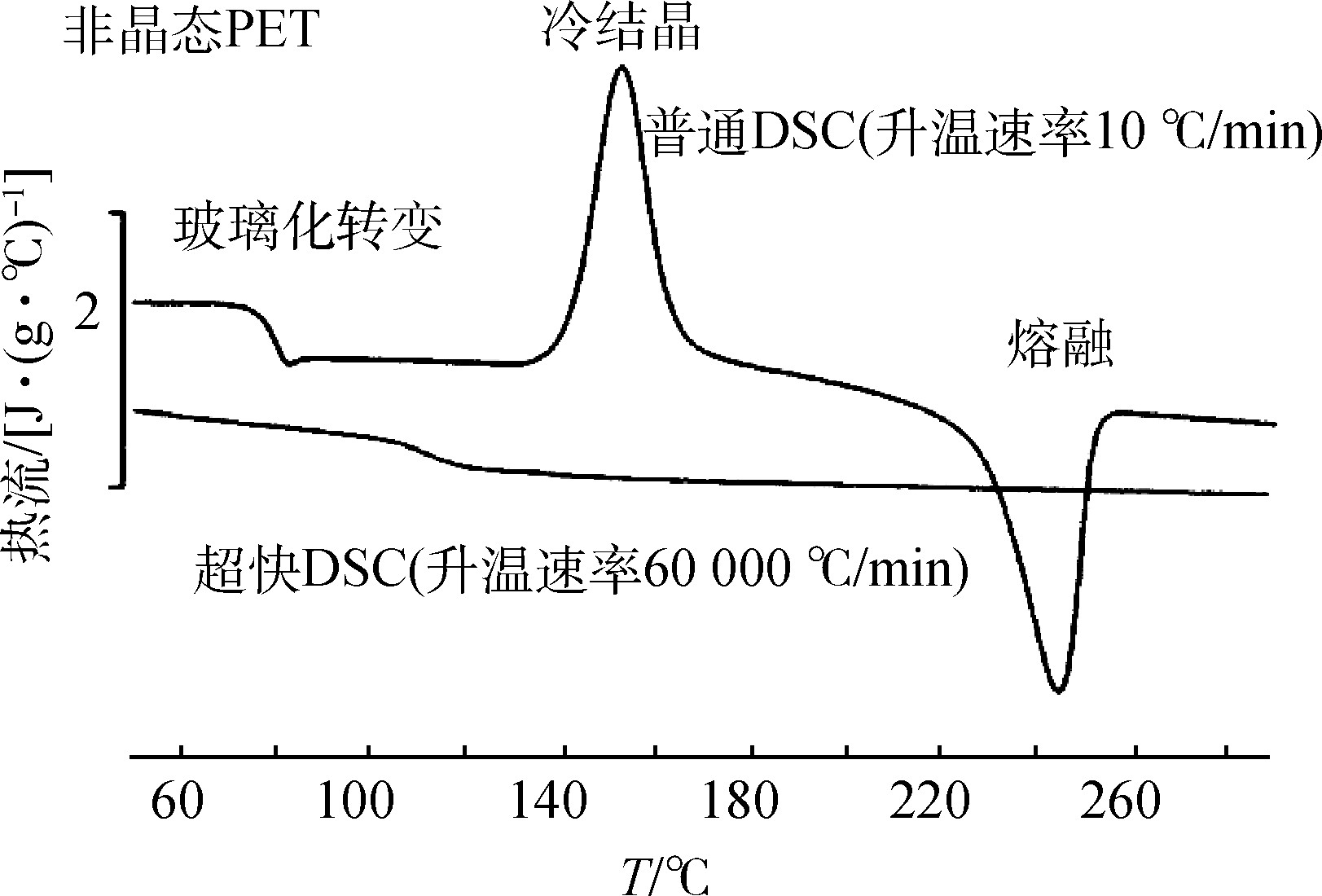
高分子材料的表征主要包括以下4个方面:形态表征,主要使用SEM、TEM、AFM等;热性能,主要使用DSC、TG等;流变性能;力学性能。其中热性能是高分子材料研究中的一个重要方面。DSC的测试曲线可以清晰地显示出高分子热力学原理中的力学性能与其热力学性质—玻璃化转化温度( Tg)、熔融温度(Tm)、结晶温度( Tc)及热焓值等具有的必然联系。氧化诱导期的测试可直接得出高分子材料的氧化行为和添加剂之间相互影响的信息。高压 DSC则可以进一步分析在不同压力测试条件下,高分子材料氧化反应、交联反应和结晶行为的变化。DSC曲线上熔融峰的形态可以反映高分子结晶物晶粒尺寸分布情况,熔融焓则直接给出了材料的结晶度[9]。在使用DSC分析检测半结晶的热塑性材料过程中,大部分的材料均会在其熔融温度前出现放热的冷结晶峰。这一现象体现出半结晶热塑性材料的收缩性,为实际生产过程中材料的选择提供了有力的帮助。例如,在传统的DSC测量中,非晶态的聚对苯二甲酸乙二酯(PET)样品在10 ℃/min的升温速率下,首先发生了玻璃化转变,然后因为原先冻结的链段被解冻后,重组发生了冷结晶;最后随着温度升高,这种冷结晶又再次被打破而发生了熔融[3]。但是在超快速DSC测量中,在60 000 ℃/min的升温速率下,非晶态的PET分子根本来不及进行重组的结晶,在热流曲线上仅观察到玻璃化转变(见图4)。DSC分析技术还可以测得杂质和湿度对高分子材料的影响。同时在冷却过程中也可反映材料的结晶温度、结晶速率、成核剂和材料加工方式之间的相互影响。二次加热曲线则直接说明加工工艺和制备条件对材料的直接影响。在高分子材料的具体应用有:聚合物共混物玻璃化转变温度的测定,依此判断共混体系的相容性,高聚物热稳定性的研究,成核剂对结晶度的影响,聚合物热老化性能研究等。
图4 传统DSC与超快速DSC对非晶态PET样品的升温DSC曲线
Fig.4 Temperature-rising DSC curves of conventional DSC and ultra-fast DSC for amorphous PET samples
4 在实验教学中的体会
许多院校为了扩大学生的知识面,开设了高分子导论等课程,并增设了一些高分子方面的实验内容。为了使这些非高分子专业学生更好地掌握DSC的工作原理及加深对DSC实验结果的理解,有必要在实验讲解中使学生清楚地理解以下几个问题。
4.1 DSC与DTA的区别及各自的应用范围
差热分析(DTA)是在程序控制温度条件下,测量样品与参比物之间的温度差与温度关系的一种热分析方法。试样在转变时热传导的变化一般是未知的,所以温差与热量变化的比例也是未知的,这使定量计算其热量变化的性能不理想。在DTA基础上增加一个补偿加热器而成的另一种技术是差示扫描量热法。DSC直接反映试样在转变时的热量变化,便于定量测定。由于各仪器的DTA/DSC曲线的吸热放热方向不同,所以曲线上必须注明吸热或放热方向。两种方法所测的转变和热效应是类似的。不同的是,DTA一般用于定性测定转变温度(峰的位置),因为温差与试样堆砌的紧密程度、传热速度和比热容等有关,用峰面积定量处理的精度较差,但DTA可测定到1 500 ℃以上;DSC适用于定量分析,因为峰面积直接对应于热效应的大小,但温度最高只能到700 ℃左右。总的来说,DSC的分辨率、重复性、准确性和基线稳定性都比DTA好,更适合有机和高分子物质的研究;而DTA更多用于矿物、金属等无机材料的分析。
用DTA方法分析上述这些反应,不反映物质的重量是否变化,也不能说明是物理变化还是化学变化,它只能反映出在某个温度下物质发生了反应,具体确定反应的实质还需要用其他方法(如光谱、质谱法等)。
4.2 高分子熔融的特点
小分子和高分子的熔融都是热力学一级相变的过程,小分子熔融过程中体系的热力学函数随温度变化范围很窄,只有0.2 ℃左右,而聚合物呈现一个较宽的熔融温度范围,即存在一个“熔限”。结晶聚合物熔融时出现熔限的原因,主要是聚合物结晶形态和完善程度不同。在结晶过程中,随温度的降低,熔体黏度迅速增加,分子链的活动性减小,在砌入晶格时来不及做充分的位置调整,使形成的晶体停留在不同的阶段;在熔融过程中,尺寸较小,比较不完善的晶体将在较低的温度下熔融,尺寸较大的、较完善的晶体需要在较高的温度下熔融,因而出现一个“熔限”;尺寸较小,比较不完善的晶体熔融后在较高的温度下进一步结晶成更完善的晶体,即所谓的二次结晶,然后再更高的温度下才能熔融。对高分子的结晶和熔融过程充分理解后,有研究者设计了这样一个实验,在测试过程中温度每升高一度,恒温24 h,在这样一个漫长的实验过程中最后终于使“熔限”变得非常窄。原因就是结晶不完善的晶体都通过二次结晶生成比较完善的晶体,然后在一个较小的温度范围内全部熔融。
4.3 高分子材料的玻璃化测试需要注意的问题
玻璃化是样品(非晶态组分)分子活动性的一个阶跃性转变[3]。材料在玻璃化转变温度之下是坚硬的,在玻璃化转变温度之上是橡胶态。玻璃化转变温度处的热容量的改变量可以用来衡量样品中非晶态组分的有序性含量。当材料退火或者长时间存储在稍低于玻璃化转变温度的情况下,随着样品向平衡态转变,会使得这种有序性得以发展。玻璃化实际上从来都是一个温度范围而不是一个确定的温度点。与玻璃化相关的分子运动是有温度依赖性和频率依赖性的,因此玻璃化转变温度随着升温速率(DSC或Dynamic mechanical analyzer, DMA)或者测试频率(DMA)的增加而提高。所以在报道玻璃化转变温度时,一定要说明测试方法(采用的是DSC或DMA)、测试条件(采用的升温速率、样品尺寸等)以及玻璃化转变温度是如何确定的(采用的1/2热容量的中点,或是拐点,或是求导后的峰值等)。玻璃化是有众多影响因素的,升温速率、升温或降温过程、老化、分子量、分子量分布、增塑剂、填料、结晶组分、共聚物、聚合物主链、侧链、氢键等,即所有影响分子链活动性的因素都影响玻璃化。
5 结语与建议
差示扫描量热法具有样品用量少、样品前处理简单、操作快速、不使用溶剂,检测结果准确度高、重现性好等优点。以上只是扼要地介绍了差示扫描量热法在高聚物研究中的一些应用,用DSC还可以做一些物理参数测定表征,研究聚合反应,高聚物热分解、低分子物的吸附与蒸发、与溶剂作用等。
在仪器技术方面, 目前发展较为迅速的是调制差示扫描量热法(Modulated differential scanning calorimetry,MDSC), 传统 DSC 只能给出总吸热或放热的热流信号, 但不能区分重叠在一起的热学现象。MDSC 是在传统的 DSC升温程序上加叠正弦调温程序, 其不仅能够得到总热流信号而且能够通过傅里叶变换, 从总热流信号中分离出可逆部分和不可逆部分, 能够更直观地看出待测物的热容等信息, 更全面地了解材料的性质[10] 。目前已有各类联用量热仪,比如红外-气相-差式扫描(Infrared spectroscopy-gas chromatography-Differential scanning calorimetry, IR-GC-DSC)联用技术及热重-差式扫描量热(Thermogravimetric analysis-Differential scanning calorimetry, TA-DSC)。联用仪器的更进一步发展会使得联用技术在未来得到更多的重视, 更丰富的物质信息会通过联用技术得到[11] 。由于热分析仪器的微量化、灵敏度、准确度、高可靠性及重复性和对环境要求的实用性,使得其在高分子领域中的应用前景更加光明。